
Mitochondrial transport and energy metabolism in synaptic transmission and nerve degeneration and regeneration
Goals and Objectives:
Mitochondria are cellular power plants that generate energy in the form of ATP to power growth, survival, and function of nerve cells. Due to their extremely polarized structures with an extended long axon, nerve cells face exceptional challenges in trafficking mitochondria to and anchoring at axons and presynaptic terminals in order to maintain energy supply. Mitochondrial trafficking and anchoring depend upon action balance of motors and anchoring proteins, so that motile mitochondria can become stationary and stationary ones can be re-mobilized. Anchored mitochondria serve as local energy sources; thus, regulation of mitochondrial trafficking and anchoring is crucial to ensure that metabolically active areas such as synapses and growth cones are adequately supplied with ATP. In addition, distal mitochondria need to be removed when they are aged or damaged throughout the neuronal lifetime. Mitochondrial dysfunction accompanied with defective transport is a key hallmark of neurodegenerative diseases. Thus, mitochondrial transport and energy metabolism in distal axons and at synapses emerge as central problems in major neurodegenerative disorders. Investigations into the regulation of mitochondrial trafficking and anchoring represent an important emerging area.
In contrast to chronic and progressive neurodegeneration, brain injury and ischemia trigger acute mitochondrial damage leading to local energy crisis. Axonal survival and regeneration requires high levels of energy consumption, and acute energy crisis contributes to regeneration failure leading to permanent neurological impairments. Thus, replacing damaged mitochondria with healthy ones will accelerate energetic recovery, and thus meet increased energy demand for nerve survival and repair. These raise a fundamental question of whether anchored mitochondria upon damaged by injury can be quickly remobilized and replaced. Our current research aims to address whether mitochondrial energetic signaling is enable neurons to enhance mitochondrial transport and restore local energy supply, thus facilitating neuronal survival and regeneration and functional recovery.
Our central hypothesis is that mitochondrial trafficking and anchoring is regulated in order to sense, integrate, and respond to changes in metabolic and growth status, synaptic activity, energy availability, pathological stress, and brain injury. Specific aims are formulated to address the following fundamental questions:
Aim 1: Elucidate mechanisms recruiting and capturing axonal mitochondria at presynaptic terminals to meet enhanced energy consumption during sustained synaptic activity,
Aim 2: Reveal an intrinsic signaling axis in mature neurons that remobilizes damaged mitochondria by turning off the anchoring switch in response to acute brain injury and ischemia and re-distributes healthy mitochondria to support nerve regeneration,
Aim 3: Determine how mature neurons maintain and recover stressed mitochondria prior to the activation of Parkin-mediated mitophagy under chronic pathological conditions,
Aim 4: Determine whether recovering local mitochondrial integrity and reversing energy metabolism facilitate CNS regeneration after injury and diseases,
Aim 5: Reveal transcellular signaling between glial cells and neurons that maintains and boosts axonal mitochondria energetic metabolism under physiological and pathological conditions.
Pursuing these investigations will elucidate important mechanisms underlying the maintenance of synaptic efficacy, nerve regeneration, and axonal mitochondrial metabolism, thus conceptually advancing knowledge of mitochondrial pathology and axonal and presynaptic energy deficits in injury and neurological disorders.
Our series of contributions to this emerging research field:
Over the past decade, systematic studies in our lab have provided mechanistic insights and conceptual advances into the regulation of axonal mitochondrial transport and anchoring and their impact on axonal energy maintenance during sustained synaptic transmission, axonal degeneration, and regeneration, as highlighted below:
- Mitochondrial trafficking and anchoring depend up the action balance of motors and anchoring proteins, so that motile mitochondria can become stationary and stationary ones can be re-mobilized. We identified syntaphilin (SNPH) as a static anchor specific for axonal mitochondria. Deleting the snph gene in mice robustly increases axonal mitochondria transport in both in vitro and in vivo nerve systems (Kang et al., Cell 2008).
- Mitochondria are essential for maintaining effective synaptic transmission by generating energy and sequesteringpresynaptic Ca2+. However, only ~33% of presynaptic terminals in the CNS retain mitochondria, and therefore sustained synaptic activity is restricted to mitochondria-containing synapses. We recently discovered a mechanistic crosstalk between presynaptic energy sensing and mitochondrial anchoring. This energy-sensitive regulation enables neurons to recruit and retain presynaptic mitochondria to maintain presynaptic energy supply, thus fine-tuning short-term synaptic plasticity and sustaining prolonged synaptic efficacy (Li et al., Nature Metabolism 2020)
- Synaptic transmission displays a notable pulse-to-pulse variation (PPV) in synaptic strength under identical stimulation. A fundamental question remains as to what presynaptic factors account for such dynamic PPV at single bouton levels. We revealed that motile axonal mitochondria dynamically passing through presynapses contribute to the PPV by fluctuating presynaptic ATP levels (Sun et al., Cell Reports, 2013).
- We revealed age-associated decline of axonal mitochondria maintenance in C. elegans; genetic manipulations that extend lifespan alter the mitochondrial maintenance profile (Morsci et al., JNS 2016).
- We demonstrated that the generation of SNPH cargo vesicles from chronically stressed mitochondria ensures that neurons respond to chronic and pathological stresses, such that dysfunctional mitochondria anchored in distal axons can be remobilized and transported to the soma for degradation (Cai et al., Current Biology 2012; Lin and Cheng et al., Neuron 2017).
- Chronic mitochondrial stress associates with major neurodegenerative diseases. Recovering stressed mitochondria and thus energy maintenance constitutes a critical step of mitochondrial quality control in early stages of neurodegeneration. We elucidated mechanisms recovering stressed mitochondria by regulating ER-mitochondrial contacts. Identifying this pathway is particularly relevant because chronic mitochondrial dysfunction and altered ER-Mito contacts have been reported in major neurodegenerative diseases (Puri et al., Nature Communications 2019).
- We investigated a fALS-linked mouse model and provided in vitro and in vivo evidence that endolysosomal deficits augment axonal mitochondrial pathology in spinal motor neurons (Xie et al., Neuron 2015).
- Mature CNS axons typically fail to regenerate after injury, leading to permanent neurological impairments. We showed that elevated SNPH expression in mature CNS neurons contributes to regeneration failure. Injury-induced acute mitochondrial damage, declined mitochondrial transport in mature neurons, and enhanced energy consumption collectively lead to local energy deficits in injured axons (Zhou et al., JCB 2016).
- We further tested our energy crisis hypothesis for regeneration failure in spinal cord injury (SCI) models. We demonstrated that enhanced axonal mitochondrial transport in snph-/-mice robustly enhanced corticospinal tract (CST) axon regeneration passing through a spinal cord lesion, accelerated regrowth of monoaminergic axons across a transection gap, and increased compensatory sprouting of uninjured CST axons. Notably, regenerating CST axons form functional synapses, transmit electrophysiological signals, and promote motor functional recovery. Thus, our study provides new mechanistic insights into intrinsic regeneration failure in the CNS and suggests that recovering local energy supply by enhancing mitochondrial transport or by boosting cellular energetics is a promising strategy to promote nerve regeneration and functional restoration after CNS injuries (Han et al., Cell Metabolism 2020).
- We further elucidated an intrinsic energetic repair signaling axis that boosts axonal energy supply by reprogramming axonal mitochondrial signaling in response to acute injury-ischemic stress in mature neurons and adult brains. Our study reveals an axonal mitochondrial signaling axis that responds to acute injury and ischemia by remobilizing damaged mitochondria for replacement, thereby maintaining local energy supply to support CNS survival and regeneration (Huang et al., Current Biology, in press).
-
Image
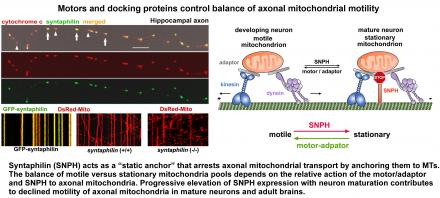
Over-expressing GFP-SNPH in WT neurons abolishes axonal mitochondrial transport. Deleting snph results in a dramatic increase in the percentage (76 ± 20%) of axonal mitochondria in mobile states relative to WT neurons (36 ± 15%). Relative motility of axonal mitochondria is controlled by action balance of motor-adaptor proteins and docking protein.




